
La recherche de nouvelles méthodes permettant de réadapter ou de combiner des médicaments existants s’est avérée un outil performant pour traiter des maladies complexes. Des médicaments utilisés pour soigner un certain type de cancer, par exemple, ont permis de renforcer efficacement des traitements ciblant d’autres cellules cancéreuses. On soigne souvent les tumeurs malignes complexes à l’aide d’une combinaison de médicaments, communément appelée « cocktail de médicaments » afin de préparer une attaque concertée contre de multiples types de cellules. Les cocktails de médicaments permettent non seulement d’éviter la résistance aux médicaments, mais également de diminuer les effets secondaires nocifs.
Cependant, il est extrêmement difficile de trouver une combinaison efficace de médicaments existants avec un dosage approprié en partie parce que les possibilités sont quasiment infinies.
Aujourd’hui, Facebook AI et le centre de recherche allemand Helmholtz Zentrum München présentent une nouvelle méthode qui va permettre d’accélérer la découverte de nouvelles combinaisons de médicaments efficaces. Nous avons créé et publié en open source le tout premier modèle d’IA qui prévoit les effets des combinaisons de médicaments, les dosages, la planification et d’autres types d’interventions telles que l’invalidation ou la suppression d’un gène. Nous avons publié en open source ce modèle baptisé CPA (Compositional Perturbation Autoencoder) comprenant une API et un pack Python très simples d’utilisation. Nous avons détaillé notre travail dans un article désormais disponible en tant que prépublication pour la communauté des chercheurs sur bioRxiv. Cet article sera également soumis à la publication dans une revue scientifique avec comité de lecture.
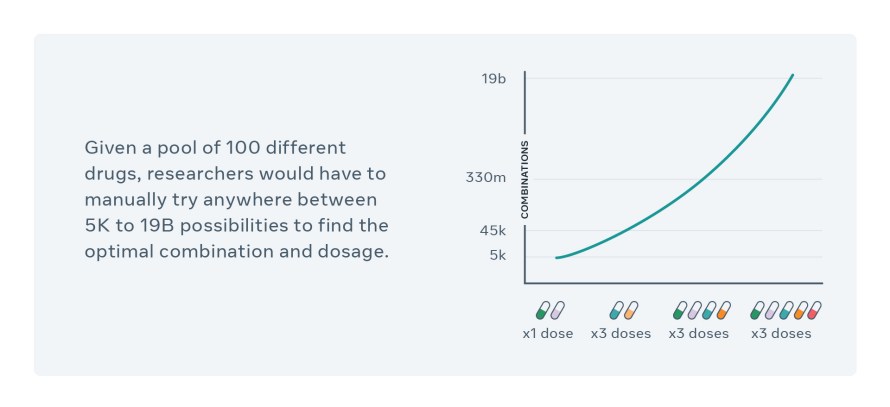
Les méthodes computationnelles permettant d’explorer de nouvelles combinaisons se sont limitées jusqu’à présent aux interactions médicamenteuses incluses dans l’ensemble de données d’apprentissage, et s’avèrent défaillantes lors du traitement de modifications moléculaires de grande dimension, comme les différents dosages et la planification. Le CPA utilise une technique d’auto-supervision innovante pour observer les cellules traitées avec une quantité limitée de combinaisons de médicaments et prévoit les effets des combinaisons inédites. Prenons un exemple simplifié : disons que les données contiennent des informations concernant l’incidence des médicaments sur différents types de cellules A, B, C et A+B. Le modèle peut découvrir l’impact individuel de chaque médicament selon le type de cellule (c’est-à-dire sur différents types de cellules), puis il recombine ces données afin d’extrapoler les combinaisons A+C et B+C ou même les interactions entre A+B et C+D.
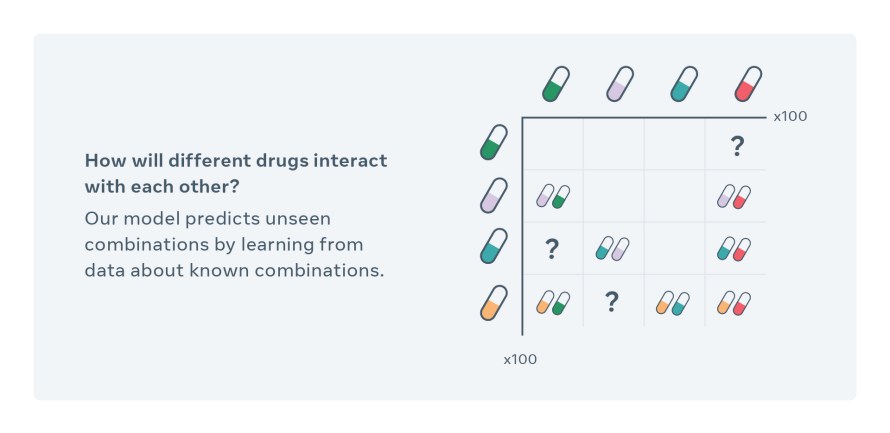
Les chercheurs pharmaceutiques peuvent utiliser le CPA pour générer des hypothèses, orienter leur processus de conception expérimental et réduire les milliards de possibilités afin de mener des expériences en laboratoire. Auparavant, il aurait fallu des années et de nombreuses expériences sur des lignées cellulaires pour tester différentes combinaisons de 100 médicaments et dosages, par exemple. Les chercheurs peuvent à présent examiner toutes les combinaisons possibles in silico (grâce à la simulation) en seulement quelques heures et sélectionner les meilleurs résultats pour s’en servir comme des hypothèses à valider et à suivre. De façon plus générale, ce travail promeut l’IA qui peut mener un raisonnement de composition ayant des implications au-delà du domaine biomédical. Par exemple, ces recherches pourraient permettre à l’IA d’améliorer sa compréhension du langage. Le raisonnement de composition donne un sens au langage et permet les représentations d’idées nuancées sur le monde qui nous entoure.
Découvrir des milliards d’interactions médicamenteuses grâce à l’auto-supervision
En biologie, le séquençage de l’ARN en cellule unique a rapidement évolué ces dernières années, générant d’énormes volumes de données avec une granularité inédite. Aujourd’hui, les chercheurs et les scientifiques utilisent le séquençage de l’ARN en cellule unique comme méthode de mesure des expressions géniques ARN des cellules individuelles au niveau moléculaire et ils étudient les effets des différentes perturbations, telles que les combinaisons de médicaments ou la suppression d’un gène, sur les systèmes biologiques.
Des chercheurs universitaires et des scientifiques ont développé et publié des ensembles de données de séquençage de l’ARN en cellule unique, portant sur des milliers, voire des millions, de cellules et comportant jusqu’à 20 000 relevés par cellule afin de faire progresser les recherches biomédicales.
De telles données de grande dimension servent de banc d’essai pour le machine learning afin de repousser les limites des prévisions combinatoires. Jusqu’ici, il n’y avait pas d’approche efficace permettant de prédire les effets des combinaisons de médicaments inédites et d’autres perturbations. Il est difficile de réaliser des prévisions à partir de ce type de données, car les modèles d’IA ont besoin d’apprendre à généraliser, voire à extrapoler, les aspects intéressants de la structure des données sans s’appuyer sur des données d’apprentissage caractérisées en vue de réaliser des prévisions portant sur de nouvelles conditions.

Pour résoudre ce problème, nous avons utilisé une technique d’apprentissage auto-supervisée nommée auto-encodage nous permettant de compresser et de décompresser les données. Ainsi, l’ordinateur est obligé de synthétiser les données pour établir des modèles de prévision utiles. Dans notre cas, l’apprentissage de l’auto-encodage est basé sur les vecteurs d’expression génique non caractérisés de différentes pathologies.
Le fonctionnement de l’apprentissage de notre modèle repose d’abord sur l’isolement des principales caractéristiques dans une cellule, comme les effets d’un médicament, d’une combinaison, d’un dosage, d’une planification, d’une suppression de gène ou d’un type de cellule spécifique. Ensuite, il recombine de manière autonome ces caractéristiques pour prévoir leurs effets sur les expressions géniques de la cellule. Pour faire une comparaison, on peut se représenter le fonctionnement du CPA en imaginant que les différentes caractéristiques des cellules (effets des médicaments, combinaisons, dosage et planification) sont des éléments vestimentaires composant la « tenue » portée par une cellule (chapeau, écharpe, lunettes ou bonnet, par exemple). Pendant la phase d’apprentissage, le CPA identifie la cellule « habillée », supprime ensuite les éléments vestimentaires pour habiller à nouveau la cellule afin d’en apprendre davantage sur tous les éléments vestimentaires et la cellule sous-jacente. Pendant la phase de test, le CPA peut prévoir les meilleures tenues (ou effets optimaux de combinaisons) : par exemple, si un chapeau et une écharpe spécifiques vont bien ensemble.
Plus concrètement, notre modèle est appliqué à un ensemble de données de séquençage de l’ARN en cellule unique concernant des cellules traitées à l’aide de différents médicaments et dosages, et son apprentissage suit les trois étapes ci-dessous :
Tout d’abord, un réseau d’encodeur transforme l’expression génique associée à une cellule dans l’ensemble de données en une « représentation de cellule. » Un réseau d’intégration traduit le traitement appliqué à cette cellule en une « représentation de traitement. » En liaison avec ce dernier, un réseau discriminateur garantit que les représentations de cellule et de traitement ne contiennent pas d’informations sur chacune d’entre elles. Ensuite, nous créons une « représentation du goulot d’étranglement » en combinant les représentations de cellule et de traitement. Enfin, un réseau décodeur traduit la représentation du goulot d’étranglement en un vecteur d’expression génique. Le modèle est entraîné de sorte que le résultat du décodeur corresponde à l’expression génique initialement disponible dans l’ensemble de données.
Dans l’étape 1 de ce processus d’apprentissage, nous estimons comment « annuler » l’effet d’un traitement pour une cellule et dans les étapes 2 et 3, nous estimons comment « appliquer à nouveau » ce même traitement à la même cellule. Bien que les traitements « annulé » et « appliqué » soient identiques pendant la phase d’apprentissage, nous intervenons dans ce processus pour utiliser notre modèle CPA afin de répondre aux questions contrefactuelles comme : « Quelle aurait été l’expression génique de la cellule si on lui avait appliqué le traitement B au lieu du traitement A ? »
Pour répondre à ces questions, nous « annulons » le traitement A à l’étape 1 et nous appliquons le traitement B aux étapes 2 et 3. Le CPA n’est pas dépendant du séquençage de l’ARN en cellule unique et il ne s’y limite pas. Il peut être facilement appliqué dans des données de séquençage de l’ARN de masse plus conventionnelles, par exemple, et aisément étendu à des relevés génomiques multimodaux.
Obtenir des prévisions fiables pour de nouvelles combinaisons de médicaments
Pour tester le CPA, nous l’avons appliqué à cinq ensembles de données de séquençage de l’ARN rendus publics contenant des mesures et des résultats pour différents médicaments, dosages et d’autres perturbations observées sur les cellules cancéreuses. Nous avons divisé chaque ensemble de données en trois catégories : apprentissage, test et hors distribution. Nous avons mesuré les performances de notre modèle à l’aide d’un indicateur R2 représentant la qualité des prévisions concernant l’expression des gènes individuels.
Sur tous les ensembles de données, les scores R2 sont restés constants entre l’apprentissage et le test. De plus, les indicateurs R2 pour les ensembles hors distribution ont été élevés. Cela signifie que les prévisions du CPA concernant les effets des combinaisons clés de médicaments et des dosages sur les cellules cancéreuses correspondent de manière fiable à celles obtenues dans l’ensemble de données de test. Pour tester les limites du modèle, nous avons réduit la quantité de données d’apprentissage et augmenté la quantité de données hors distribution. Malgré une diminution considérable des performances, les prévisions étaient loin d’être aléatoires. Le modèle n’est pas conçu pour une utilisation dans des scénarios si extrêmes, bien qu’il soit intéressant de cerner ses capacités d’apprentissage.
Perspectives d’avenir
Nous espérons que les biologistes ainsi que les chercheurs de l’industrie pharmaceutique et universitaires s’appuieront sur cet outil open source afin d’accélérer le processus d’identification des combinaisons optimales de médicaments pour différentes maladies. Ses API et son pack Python prêts à l’emploi sont conçus pour aider les chercheurs à se connecter à des ensembles de données et à examiner des prévisions sans aucune compétence en machine learning.
En offrant aux laboratoires pharmaceutiques des outils assistés par l’IA, nous espérons accélérer considérablement le processus d’identification des combinaisons optimales de médicaments et d’autres interventions, ce qui pourrait aboutir à l’élaboration de meilleurs traitements pour les maladies complexes comme le cancer et les nouvelles maladies comme le COVID-19.
À plus long terme, le CPA ouvre la voie à de toutes nouvelles possibilités en matière de développement de traitements médicamenteux. À l’avenir, il pourrait non seulement accélérer les recherches sur la réaffectation des médicaments, mais aussi permettre un jour la personnalisation des traitements en fonction des réponses des cellules individuelles, un des enjeux les plus importants pour les progrès de la médecine.