
Finding new ways to repurpose or combine existing drugs has proved to be a powerful tool to treat complex diseases. Drugs used to treat one type of cancer, for instance, have effectively strengthened treatments for other cancer cells. Complex malignant tumors often require a combination of drugs or “drug cocktails,” to formulate a concerted attack on multiple cell types. Drug cocktails can not only help stave off drug resistance but also minimize harmful side effects.
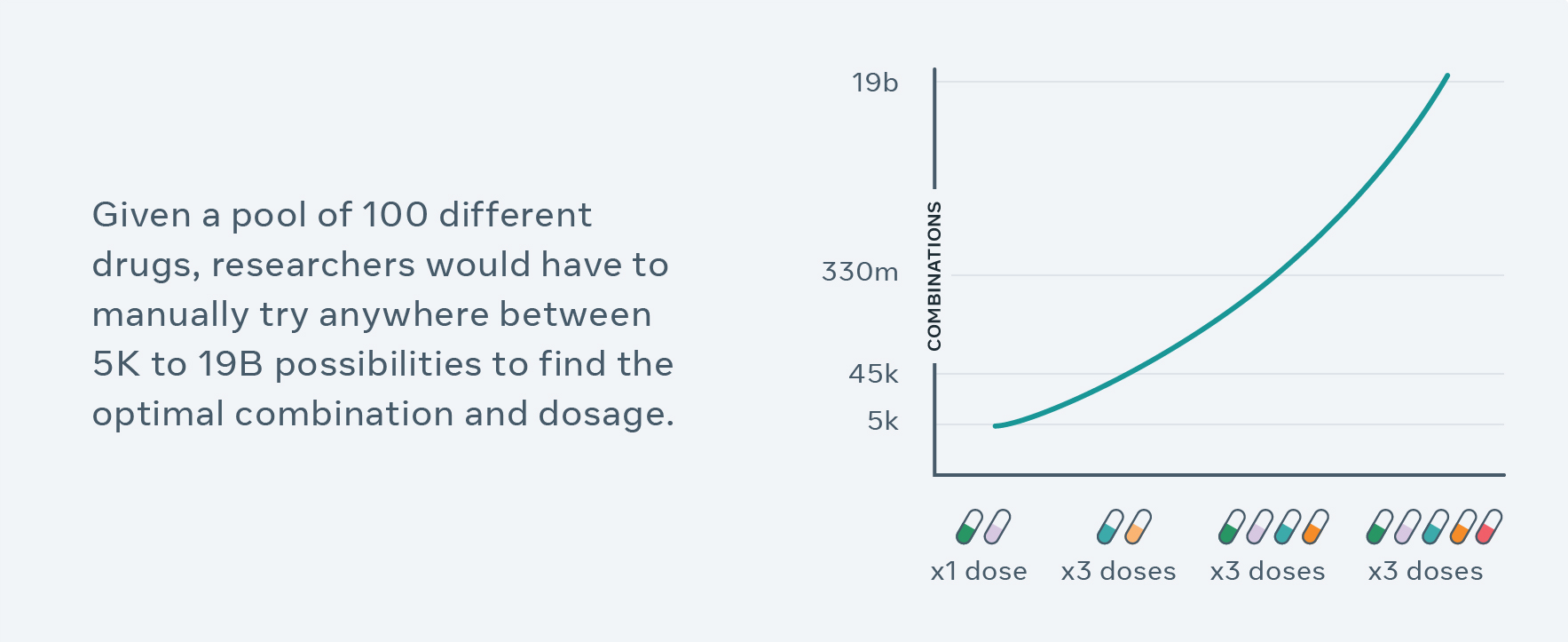
But finding an effective combination of existing drugs at the right dose is extremely challenging, partly because there are near-infinite possibilities.
Today, Facebook AI and the Helmholtz Zentrum München are introducing a new method that will help accelerate discovery of effective new drug combinations. We’ve built the first single AI model that predicts the effects of drug combinations, dosages, timing and even other types of interventions, such as gene knockout or deletion. We’re open-sourcing this model, called Compositional Perturbation Autoencoder (CPA), including an easy-to-use API and Python package. We have detailed the work in a paper now available to the research community as a preprint on bioRxiv. The work will also be submitted to a peer-reviewed journal.
Computational methods for exploring new combinations so far have been limited to drug interactions included in the training data set and break down when handling high-dimensional molecular-level changes, such as different doses and timing. CPA uses a novel self-supervision technique to observe cells treated with a finite number of drug combinations and predicts the effect of unseen combinations. For a simplified example, let’s say that data contains information about how drugs affect different types of cells A, B, C and A+B. Now the model can learn the individual impact of each drug in a cell-type specific fashion (i.e., on different types of cells) and then recombine them in order to extrapolate combinations of A+C, B+C, or even how A+B interacts with C+D.
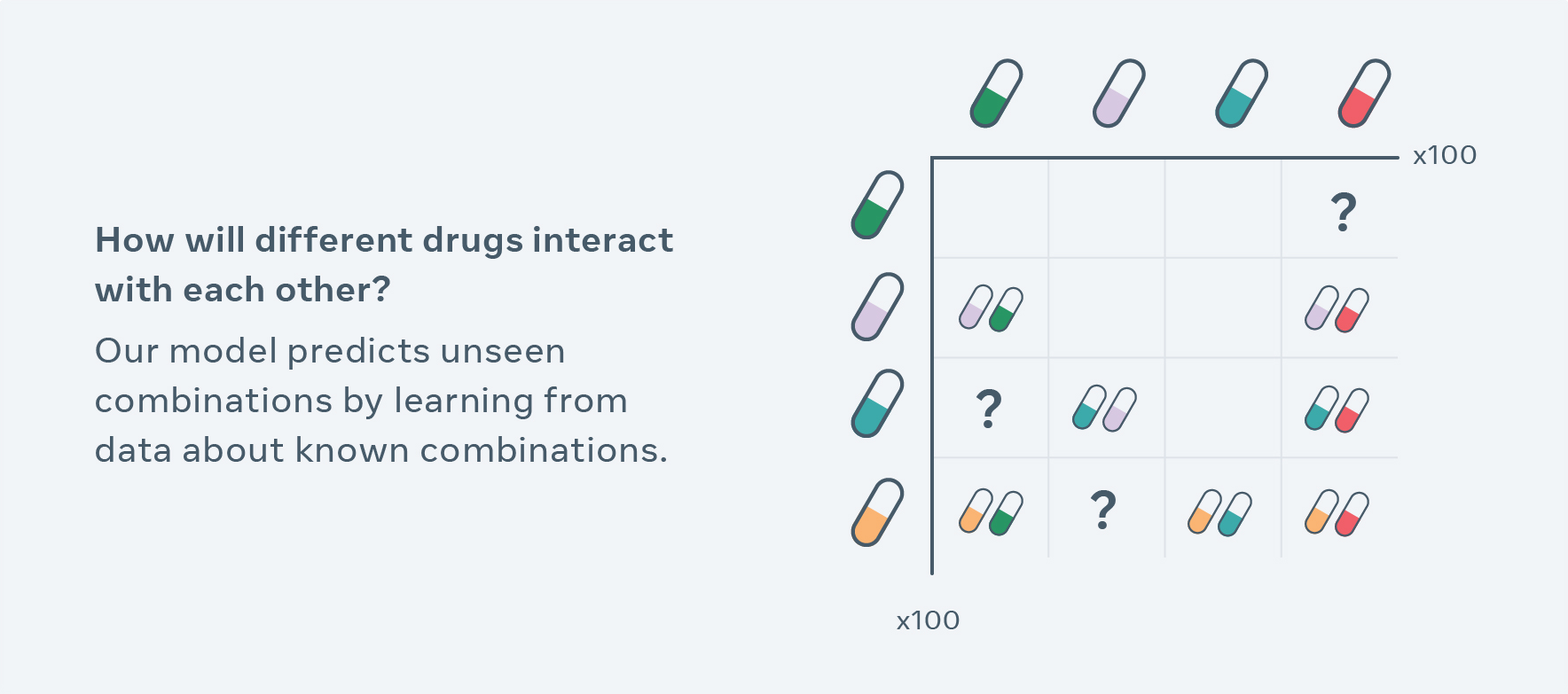
Pharmaceutical researchers can use CPA to generate hypotheses, guide their experimental design process and help narrow down billions of choices to run experiments in the lab. For example, previously, it would typically take years and many cell-line experiments to test different combinations of 100 drugs and doses — researchers can now screen all possible combinations in silico (in simulation) within just a couple hours, and select top results as hypotheses for validation and follow-up. More broadly, this work advances AI that can do compositional reasoning, which has implications beyond biomed and could lead to AI that can better understand, for instance, language. Compositional reasoning gives meaning to language and makes representations of nuanced ideas about the world.
Learning Billions of Drug Interactions With Self-Supervision
In biology, single cell RNA sequencing has been rapidly evolving in recent years, producing vast amounts of data with more granularity than ever before. Researchers and scientists today use single-cell RNA sequencing as a way to measure RNA gene expressions of individual cells at the molecular level and study the effects of various perturbations, such as drug combinations or gene deletion, on the biological systems. Academic researchers and scientists have built and released public single-cell RNA sequencing data sets — composed of thousands to millions of cells with as many as 20,000 readouts per cell — to facilitate biomedical research.
Such high-dimensional data provides a strong test bed for machine learning (ML) to push the boundaries of combinatorial predictions. So far, there hasn’t been an effective approach to predict the effects of unseen drug combinations and other perturbations. Making predictions from this type of data is challenging because AI models need to learn how to generalize, or even extrapolate, interesting aspects of the structure of data — without relying on labeled training data — to make predictions about new conditions.

To solve this, we used a self-supervised learning technique called auto-encoding, in which we “compress” and “decompress” data, forcing the machine to essentially summarize the data into useful patterns for prediction. In our case, the auto-encoder learns from unlabeled gene expression vectors from different conditions.
Our model works by first separating and learning about key attributes in a cell, such as effects of a certain drug, combination, dosage, time, gene deletion or cell type. Then, it independently recombines the attributes to predict their effects on the cell’s gene expressions. As an analogy, one way to think about how CPA works is to imagine the different cell attributes, such as effects of drugs, combinations, dosage and timing, as clothing items that make up an “outfit” the cell is wearing, like hats, scarves, glasses and beanies. In training, CPA sees the dressed-up cell, removes articles of its clothing, and redresses the cell to learn about all the clothing and the underlying cell. During testing, CPA can then predict the best outfits (or optimal effects of combinations) — how a hat would look with a particular scarf, for example.
More specifically, our model takes a single-cell RNAseq data set of cells treated with several drugs and doses and then learns:
- First, an encoder network transforms the gene expression associated to one cell in the data set into a “cell representation.” An embedding network translates the treatment applied to this cell into a “treatment representation.” In tandem, a discriminator network ensures that cell and treatment representations do not contain information about each other.
- Second, we construct a “bottleneck representation” by combining the cell and treatment representations.
- Third, a decoder network translates the bottleneck representation back into a gene expression vector. The whole model is trained such that the output of the decoder matches the gene expression initially available in the data set.
In step 1 of the previous training process, we estimate how to undo the effect of a treatment of a cell, and in steps 2 and 3, we estimate how to reapply that same treatment to the same cell. Although the undone and applied treatments are the same during training, we intervene in this process and use our CPA model to answer counterfactual questions, such as “What would have been the gene expression of this cell, had it been treated with treatment B instead of treatment A?”
To answer these questions, we undo treatment A in step 1, and apply treatment B in steps 2 and 3.
CPA is not dependent or restricted to single cell RNA sequencing alone. It can be easily applied in the more traditional bulk RNAseq data as well, for instance and readily extended to multimodal genomics readouts.
Achieving Reliable Predictions of Novel Drug Combinations
To test CPA, we applied it on five publicly available RNA sequence data sets that contain measurements and outcomes of various drugs, doses and other perturbations on cancer cells. We split each data set into training, testing and out-of-distribution (OOD). We measured the performance of our model in terms of the R2 metric, which represents how well we predict the expression of individual genes.
On all of the data sets, the R2 scores stayed consistent between the training and testing, and the R2 metrics for OOD were high as well. Meaning, CPA’s predictions of the effects of key drug combinations and doses on cancer cells reliably matched those found in the testing data set. To further test the limits of the model, we reduced the amount of training data and increased the amount of OOD data. While the performance dropped substantially, the predictions were still significantly above random. The model is not intended to use in such extreme scenarios, though it’s interesting to understand its learning capabilities.
Looking Ahead
Our hope is that pharmaceutical and academic researchers as well as biologists will utilize this open source tool to accelerate the process of identifying optimal combinations of drugs for various diseases. Its ready-to-use APIs and Python package are designed to make it easy for researchers to plug in data sets and run through predictions without ML expertise.
By providing pharmaceutical labs with AI-powered tools, we hope to help dramatically accelerate the process of identifying optimal combinations of drugs and other interventions that could ultimately lead to better treatments for complex diseases like cancer and novel diseases like COVID-19.
Looking further ahead, CPA paves the path for entirely new opportunities in developing drug treatments. In the future, it could not only speed up drug repurposing research, but also — one day — make treatments much more personalized and tailored to individual cell responses, one of the most active challenges in the future of medicine to date.


